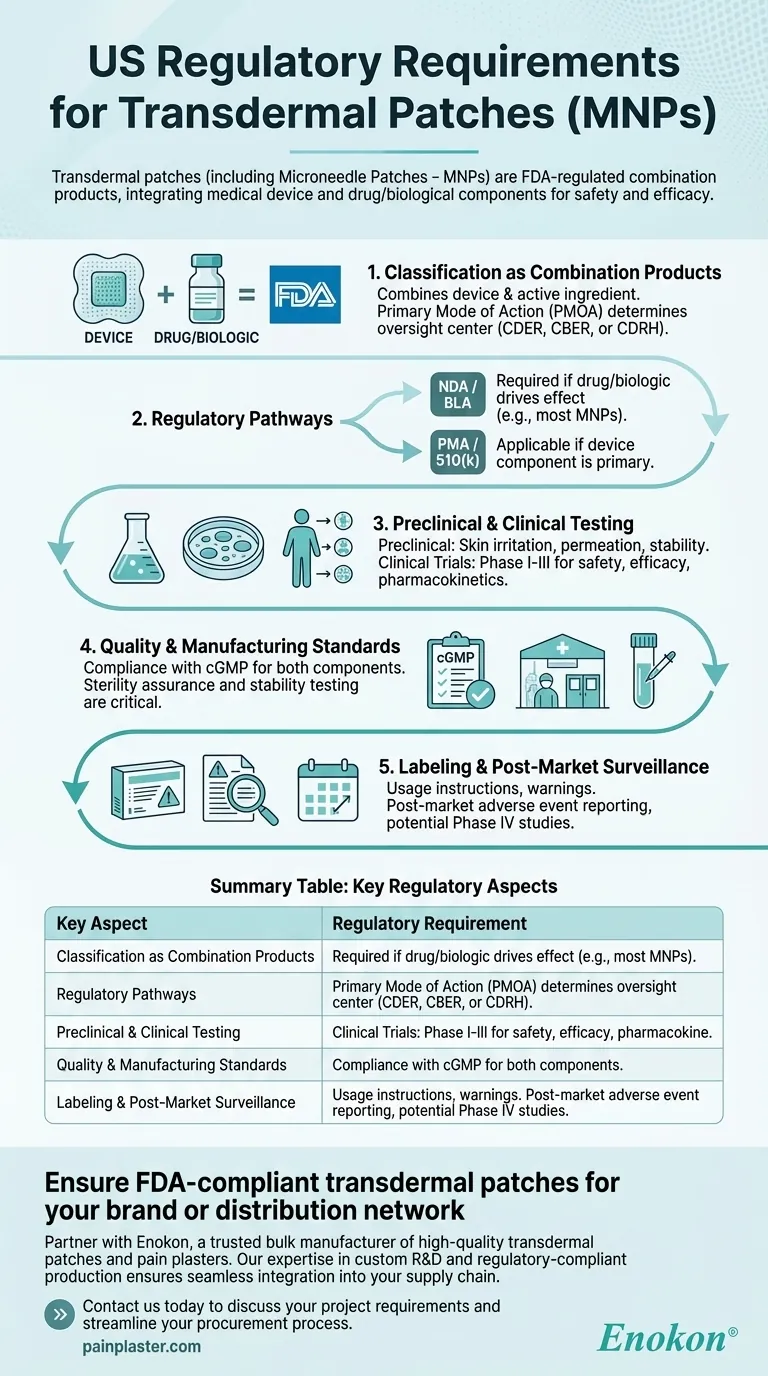
Os adesivos transdérmicos, incluindo os adesivos com microagulhas (MNPs), são regulamentados nos EUA como produtos combinados pela FDA, exigindo processos de aprovação rigorosos para garantir a segurança e a eficácia. Estes produtos integram componentes de dispositivos médicos e medicamentos/biológicos, exigindo a conformidade com vias regulamentares específicas, dependendo do seu modo de ação primário. O processo de aprovação envolve testes pré-clínicos e clínicos, controlo de qualidade e cumprimento de normas de rotulagem e fabrico.
Pontos-chave explicados:
-
Classificação como produtos combinados
- A FDA classifica os adesivos transdérmicos como produtos combinados porque combinam um dispositivo médico (por exemplo, suporte do adesivo, microagulhas) com um medicamento ou ingrediente ativo biológico.
- O modo de ação primário (PMOA) determina se o produto é regulado pelo Centro de Avaliação e Investigação de Medicamentos (CDER), pelo Centro de Avaliação e Investigação Biológica (CBER) ou pelo Centro de Dispositivos e Saúde Radiológica (CDRH).
-
Vias de regulamentação
- Pedido de Novo Medicamento (NDA) ou Pedido de Licença Biológica (BLA): Necessário se o fármaco ou o componente biológico for responsável pelo efeito terapêutico.
- Aprovação prévia à comercialização (PMA) ou 510(k): Aplicável se o componente do dispositivo for primário (por exemplo, microagulhas que facilitam a administração do medicamento).
- Os MNP seguem frequentemente a via NDA/BLA devido à sua função centrada no medicamento.
-
Ensaios pré-clínicos e clínicos
- Estudos pré-clínicos: Incluem testes de irritação cutânea, permeação e estabilidade para avaliar a segurança e a eficácia da administração.
- Ensaios clínicos: Os ensaios de fase I-III avaliam a farmacocinética, a eficácia e os efeitos adversos em seres humanos.
-
Normas de qualidade e fabrico
- Conformidade com as actuais Boas Práticas de Fabrico (cGMP) para componentes de medicamentos e dispositivos.
- A garantia de esterilidade e os testes de estabilidade são fundamentais para os adesivos com produtos biológicos.
-
Rotulagem e vigilância pós-comercialização
- Os rótulos devem incluir instruções de utilização, avisos e condições de armazenamento.
- Os requisitos pós-comercialização incluem a comunicação de eventos adversos e potenciais estudos de Fase IV.
Para os compradores, a compreensão destes requisitos garante o alinhamento com fornecedores em conformidade com a FDA, reduzindo os riscos de produtos não conformes. Já considerou como estes regulamentos afectam os seus prazos de aquisição ou critérios de seleção de fornecedores? A interação entre a supervisão de dispositivos e medicamentos molda discretamente a fiabilidade das terapias transdérmicas modernas.
Tabela de resumo:
| Aspeto-chave | Requisito regulamentar |
|---|---|
| Classificação | Regulamentados como produtos combinados (dispositivo + medicamento/biológico) pela FDA. |
| Modo de ação primário | Determina a supervisão pelo CDER (medicamento), CBER (biológico) ou CDRH (dispositivo). |
| Vias de aprovação | NDA/BLA (orientada para o medicamento) ou PMA/510(k) (orientada para o dispositivo). Os MNP seguem normalmente a NDA/BLA. |
| Requisitos de ensaio | Ensaios pré-clínicos (segurança, permeação) e clínicos (Fases I-III). |
| Normas de fabrico | Conformidade com as cGMP para componentes de medicamentos e dispositivos; garantia de esterilidade para produtos biológicos. |
| Obrigações pós-comercialização | Comunicação de eventos adversos, estudos de Fase IV e conformidade com a rotulagem. |
Garanta adesivos transdérmicos em conformidade com a FDA para a sua marca ou rede de distribuição
Faça parceria com a
Enokon
um fabricante de confiança de adesivos transdérmicos e pensos analgésicos de alta qualidade. A nossa experiência em I&D personalizada e produção em conformidade com a regulamentação garante uma integração perfeita na sua cadeia de fornecimento.
Contacte-nos hoje
para discutir os requisitos do seu projeto e simplificar o seu processo de aquisição.
Guia Visual
Produtos relacionados
- Patch de alívio da dor em gel de mentol
- Patch de alívio da dor Icy Hot Menthol Medicine
- Pensos de aquecimento para aliviar as dores das cólicas menstruais
- Adesivo para o alívio da tosse e das dores provocadas pela asma para adultos e crianças
- Patch antidiarreico medicado com ervas para alívio digestivo
As pessoas também perguntam
- Qual a eficácia dos pensos analgésicos para as dores musculares?Alívio direcionado sem efeitos secundários sistémicos
- Quem deve consultar um profissional de saúde antes de utilizar pensos para alívio da dor?Guia de segurança
- O que são adesivos para alívio da dor e como funcionam?Descubra o tratamento não invasivo da dor
- Quando é que deve considerar a utilização de produtos para o alívio da dor, como cremes e adesivos?Otimizar o conforto e a segurança
- Quais são algumas das aplicações comuns dos pensos para alívio da dor?Alívio direcionado para o controlo da dor